Медичний термін "фібриляція", напевно, пам'ятають усі, хто дивився телевізійний серіал "Швидка допомога". За констатацією цього невідомого простому смертному стану пацієнта слідувала команда: "Розряд!" - після чого лікарі повідомляли про настання дефібриляції, і серце відновлювало свою діяльність. Що ж таке фібриляція? Це стан, при якому клітини серцевого м'яза - кардіоміоцити - скорочуються неузгоджено, асинхронно. В результаті серце як насос не виконує своєї функції: кров не циркулює по судинах і кисень не надходить до клітин організму. Це кисневе голодування особливо небезпечно для мозку - він живе в таких умовах лише кілька хвилин.
Смертність людей від різних захворювань серця займає сумне перше місце. Один із наслідків серцевих недуг і є фібриляція, причини виникнення якої досліджуються в усьому світі, але все ще так і не встановлені. Ми намагаємося зрозуміти механізм цього виду аритмій з 1985 р і, як нам представляється, близькі до його розкриття. Про результати багаторічної роботи і піде мова.
У попередній статті ми розповіли про новий тип іонних каналів, відкритому приблизно 15 років тому [ 1 ]. Тепер ми переконалися, що саме ці канали мають пряме відношення до фібриляції.
Механіка, електрика і аритмії
Функція серця, як відомо кожному, - забезпечувати циркуляцію крові по судинах. Відбувається це завдяки послідовному розслабленню передсердь і шлуночків (фаза діастоли), а потім їх скорочення (фаза систоли). Закінчується цикл загальним розслабленням всіх чотирьох камер. Скорочення серцевого м'яза (міокарда) викликається електричним імпульсом, який виникає в клітинах синусно-передсердного вузла, званого водієм ритму. Далі збудження передається по передсердям і досягає передсердно-шлуночкового вузла, через пучок Гіса доходить до його кінцевих розгалужень - волокон Пуркіньє - і передається шлуночків. Клітини серцевих камер скорочуються і розслабляються узгоджено, і тим забезпечується чіткий ритм в діяльності насоса, переганяють кров.
Незбуджені клітини міокарда мають потенціал спокою (від -70 до -90 мВ), який під дією різних стимулів змінюється. Він може стати більш негативним, тоді мембрана гиперполяризуется, але може і зменшитися при її деполяризації. Вхідні в клітку катіони (перш за все Na + і Ca 2+) завжди деполярізуют мембрану, а виходять (в основному K +) - гіперполяризуючий. Швидкий зрушення потенціалу спокою в позитивному напрямку називають потенціалом дії (ПД). У ньому прийнято виділяти три фази: деполяризації, овершута (перескоку) і реполяризації. Під час першої фази потенціал стрімко наростає і мембрана втрачає свій нормальний заряд; у другій фазі потенціал набуває позитивне значення, а протягом третьої повертається до початкової величини - потенціалу спокою. Саме потенціал дії і спонукає клітини серця до скорочення.
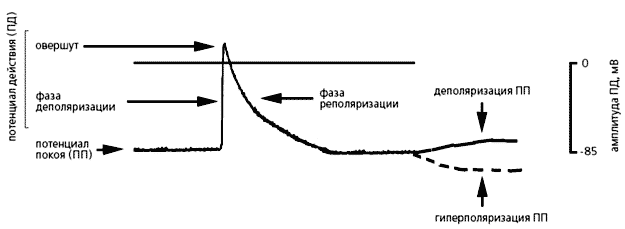
Фази потенціалу дії.
Пряма залежність скорочення кардіоміоцитів від її порушення (виникнення ПД), тобто електромеханічне спряження, вже детально вивчена. Але існували клінічні дані, що свідчать також про наявність в міокарді зворотного зв'язку - зміні електричних процесів під дією механічних факторів: розтягування міокарда і (або) зміни його скорочувальної активності. Така залежність фактично була встановлена ще в 1915 р англійським кардіофізіологом Ф.А.Бейнбріджем. Він показав, що розтягнення правого передсердя викликає почастішання ритму серця у щурів. Співвітчизники Бейнбріджа О.Франк і Е.Х.Старлінг, розтягуючи серцевий м'яз до певної довжини, домоглися збільшення сили її скорочення. З цього можна було б зробити висновок, що механічна стимуляція тканини серця повинна приводити до зміни мембранного потенціалу в клітинах. Однак до недавнього часу вплив зворотного зв'язку не вивчалось і не враховувалося. Про її існування в 1968 р лондонський фізіолог М.Дж.Лаб висловив лише припущення, але не довів його через недосконалість експериментальних методів.
До теперішнього часу накопичилося безліч підтверджують цю думку клінічних спостережень. Добре відомі, наприклад, передсердні аритмії - фібриляція, пароксизмальнатахікардія, - які виникають у хворих з гострим розтягненням передсердь або при поступовому збільшенні їх розмірів. Описані випадки, коли механічно индуцированная аритмія виникала у пацієнтів і без цих патологій, просто при введенні катетера в серце. До різних порушень серцевого ритму можуть призводити також постійні механічні перевантаження, викликані на артеріальну гіпертензію, застійної серцевої недостатністю і хронічним розтягненням міокарда. Взаємозв'язок механіки і електрики підтверджується і давно прийнятої в медицині механічної стимуляцією серця. Цей непрямий масаж успішно застосовують, щоб відновити скорочення міокарда або вивести його зі стану фібриляції.
Таким чином, клінічні спостереження ілюструють, що розтягнення камер серця, особливо при його патології, призводить до аритмій, але механічна стимуляція може і відновлювати нормальний серцевий ритм, запобігати розвитку фібриляції. Тому надзвичайно важливо зрозуміти, завдяки яким процесам здійснюється механоелектричного зворотний зв'язок.
Робота кардіоміоцитів
У фізіологічних умовах під час розслаблення серцевих камер (тобто в діастолі) механічний вплив на міокард призводить до зміни потенціалу спокою на мембрані кардіоміоцитів і отже протікає через неї струму. Ця відповідь обумовлений роботою механочувствітельние іонних каналів (в науковій літературі їх називають механосенсітівнимі і позначають абревіатурою МСК, яку ми збережемо в даному тексті).
Залежність електричної активності кардіоміоцитів від механічних впливів на них ми почали вивчати давно. В експериментах імітували нормальні скорочення цих клітин в залежності від величини розтягування тканини, для чого розтягували фрагменти тканини міокарда з силою, рівною фізіологічної, а потім перевищували цю силу і вимірювали мембранний потенціал. В результаті з'ясувалося, що додаткове розтягнення (1.5 мН) тканини правого передсердя призводить до зміни звичайної форми потенціалу дії: в фазі реполяризації, на рівні 90%, починалася деполяризація мембрани. Якщо розтягнення продовжували, виникав позачергової потенціал дії. У дослідах на тканини передсердь, взятої від тваринного, який переніс інфаркт лівого шлуночка, досить було перевищити силу розтягування над фізіологічним значенням всього на 0.1 мН, щоб викликати більш серйозні зміни: збільшувалася тривалість реполяризації вже на двох рівнях (50 і 90%); вдвічі більша сила приводила до додаткового потенціалу дії; якщо ж вона досягала 0.3 мН, починалася фібриляція передсердя [ 2 ]. Схожі результати дали експерименти на фрагментах шлуночків серця [ 3 ].

Отже, при розтягуванні міокарда або зміні сили скорочень спочатку настає деполяризація мембрани і змінюється тривалість потенціалу дії головним чином за рахунок аномально протікає реполяризації (на різних її рівнях). Потім на цій же фазі може початися повторна деполяризация, в результаті чого порушується нормальний ритм серця - виникають аритмії, аж до фібриляції [ 4 ].

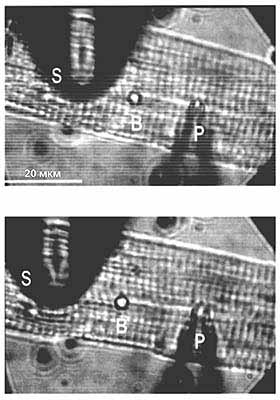
Мікрофотографії ізольованого кардиомиоцита. Вгорі: клітина, до мембрани якої підведені дві микропипетки - реєструє (Р) і розтягує (S), - але жодна ще не введена в дію внизу - розтягнутий кардіоміоцит. Для візуального спостереження за розтягуванням між пипетками поміщений гумову кульку (B). Через першу піпетку, яка містить електроліт, подається негативний тиск такої сили, щоб прорвати мембрану і встановити контакт між вмістом клітини і електролітом, після чого і реєструється іонний струм. Другий піпеткою клітина тільки розтягується. За результатами вимірювання сили струму до і після механічного впливу можна судити про вплив останнього на генерацію іонного струму клітинами серцевого м'яза.
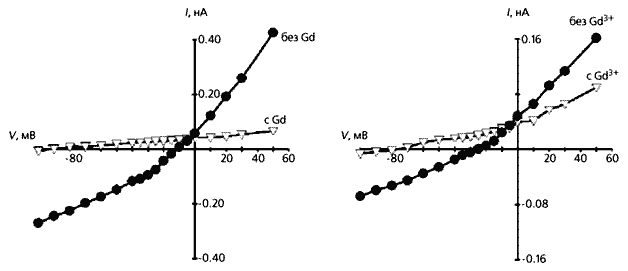
Оскільки ефект розтягування тканини міокарда блокувався іонами гадолінію - інгібіторами механочувствітельние каналів, - напрошувався висновок про їхню участь в описаних подіях [ 5 ]. Але щоб довести це, необхідно було дослідити іонні струми і самі МСК нема на тканини серця, а на окремих життєздатних клітинах. Для розтягування одиничного кардиомиоцита і вимірювання в ньому струму, ми розробили спеціальний прийом, який і використовували в подальших експериментах на ізольованих клітинах з шлуночків серця [ 4 ].
Результати виявилися подібними з тими, що були отримані на тканини передсердь: чим з більшою силою розтягувалася клітина, тим значніше для неї були наслідки; кардіоміоцити хворого серця виявилися більш чутливими до розтягування, ніж здорового, незалежно від того, були це клітини морської свинки або людини; сильніше, у порівнянні з кардиомиоцитами здорової щури, відповідали на розтягнення клітини тварини, яка страждає на артеріальну гіпертензію. Сила відповіді залежала також і від віку: для змін величини потенціалу спокою і тривалості потенціалу дії потрібно набагато більше розтягання клітин серця молодих щурів, ніж старих.
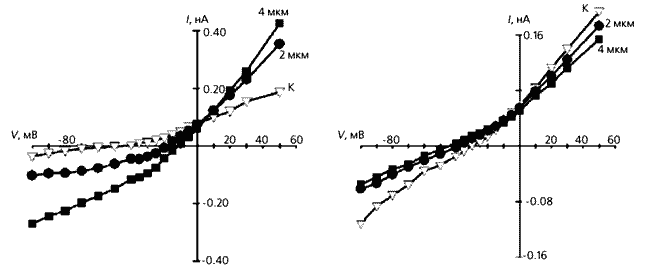
Зміна сили неселективного катіонного струму і потенціалу його реверсії в кардіоміоциті з лівого шлуночка людського серця після інфаркту міокарда. Вгорі - загальний струм до механічного впливу на клітину (1) і після її розтягування на 4 мкм (2). Видно, що в розтягнутій клітці змінюється як сила струму, так і потенціал, при якому виходить з клітки ток змінюється на вхідний: зменшується за абсолютною величиною з -60 мВ приблизно до -20 мВ. Внизу - струм, що протікає через розтягнутий кардіоміоцит з додаванням іонів гадолінію (Gd) і без них. Тут проявляється роль цих іонів як інгібіторів механочувствітельние каналів.
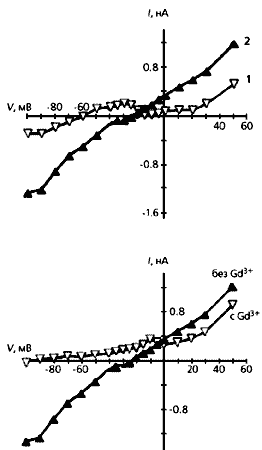
Таким чином, пряме розтягування як передсердних, так і шлуночкових кардіоміоцитів викликає деполяризацію їх мембрани і збільшення фази реполяризації на рівні 90%. Якщо обидва ці процеси досягають критичних величин, виникає додатковий потенціал дії, що призводить до екстрасистол - позачерговому скорочення серцевих камер.
Сумарний струм, що протікає через мембрану кардіоміоцитів, теж збільшувався в міру розтягування клітини і зникав при поверненні її довжини до вихідної. Якими іонами викликаний ток, ми встановили в експериментах, замінюючи один іон на інший, наприклад K + на Cs +, Na + на більший катіон тріетіламмонія, а Cl - на F -. В результаті з'ясувалося, що при розтягуванні клітини за струм відповідальні іони натрію, що входять до неї через механочувствітельние канали. У дослідах по дослідженню струмів ізольованих кардіоміоцитів використовувалися зразки, взяті з тих же об'єктів, тобто здорових морських свинок і щурів (молодих і старих), людей із захворюваннями серця і щурів з гіпертензією. І тут картина була такою ж, як в експериментах з вивчення потенціалу дії: чутливість клітин до експериментального розтягування збільшувалася з віком тварин, але ще сильніше була пов'язана з гіпертензією. Таке підвищення чутливості ми пояснюємо збільшенням на мембрані числа механочувствітельние каналів [ 6 ].
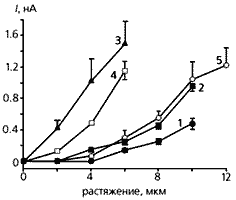
Залежність сили струму від величини розтягування шлуночкових кардіоміоцитів з серця щура (1-3), хворої людини (4) і тримісячної морської свинки (5). Як видно з графіка, найменше реагує на таке механічний вплив клітина молодий щури (1), кілька сильніше - старої (2), і найбільш сильний електричний відповідь кардиомиоцита у щура того ж віку, але хворий гіпертензією (3); реакція ж клітини з хворого серця людини трохи нижче, ніж у такий щури, а клітина міокарда молодий морської свинки відповідає на розтягнення струмом майже тієї сили, що кардіоміоцит старої щури.
Отже, отримані нами результати дозволяють зробити висновок, що не тільки електричне збудження призводить до скорочення або розслаблення клітин серця, тобто до їх механічного зміни, а й навпаки - на механічний вплив кардіоміоцити відповідають електричною активністю. Однак якщо прямий зв'язок забезпечує нормальний серцевий ритм, то зворотна позначається протилежним чином - порушенням ритміки при всіх патологіях.
Вважається, що механічні подразнення сприймаються лише кардиомиоцитами - Електровозбудімость скорочувальними клітинами. Ми ж довели, що в зворотного зв'язку, особливо при захворюваннях серця, можуть брати участь не тільки вони, а ще й фібробласти - нем'язові електроневозбудімие клітини.
Нова роль фібробластів
У нормальному міокарді група нем'язові клітин складається з різних типів, але переважають в ній фібробласти, яким приписується роль опорних структур. Однак, судячи з нагромадженням відомостями про інші сторони метаболічної активності фібробластів, вони беруть участь і в регуляції роботи серця [ 4 , 7 ]. До такого припущення ми прийшли ще в 1986 р Вивчення електрофізіологічних характеристик фібробластів серця, що б'ється і їх міжклітинної взаємодії привело нас до думки, що діяльність міокарда забезпечується не одними лише кардиомиоцитами, а їх спільним функціонуванням з цими опорними клітинами. У такому випадку робота і здорового серця, і зміненого хворобою повинна виглядати інакше, ніж прийнято вважати.
В серці ссавців не так багато фібробластів, приблизно 5-10%, але в зоні синусового вузла їх кількість досягає 45-75%. Ці нем'язові клітини відрізняються від кардіоміоцитів цілим рядом електрофізіологічних особливостей. До їх числа відносяться низька величина потенціалу спокою (в середньому -25 мВ) і високий вхідний опір - близько 1 ГОм. У ритмі спонтанних або викликаних скорочень міокарда фібробласти генерують механоіндуцірованние потенціали (МІП), форма яких абсолютно не схожа на форму потенціалу дії кардіоміоцитів. Амплітуда МІП фібробластів або порівнянна з величиною потенціалу спокою кардіоміоцитів, або значно менше його. Характерно, що МІП ніколи не перескакує через нульове значення, тобто в його формі відсутній овершут. В експериментах величина потенціалу реверсії залежить від ступеня розтягування препарату, але при стандартній силі впливу вона становить всього -5 мВ. В однакових експериментальних умовах механоіндуцірованний потенціал фібробластів генерується пізніше, ніж потенціал дії кардіоміоцитів: в серці жаби на 90 мс, щури - на 10, людини - на 40 мс. За тривалістю МІП збігається з часом скорочення препарату.
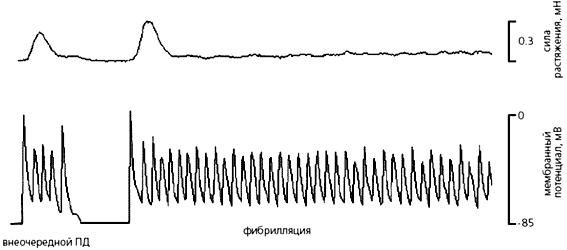
Праворуч - механограмму (вгорі) і потенціали, що виникають в кардіоміоциті (в середині) і фібробластів (внизу) правого передсердя щура. Видно, що потенціал дії (ПД) кардіоміоцити призводить до скорочення тканини серця, а механоіндуцірованний потенціал (МІП) фибробласта з'являється при стисканні цієї клітини під час скорочення тканини.
У нормальних фізіологічних условиях МІП фібробластів вінікає у відповідь на СКОРОЧЕННЯ серця. У период систоли фаза наростання цього потенціалу пов'язана з актівацією неселективних механочувствітельніе каналів, через Які в клітку надходять Різні іоні, в тому чіслі Ca 2+. Если збільшується концентрація іонів кальцію, что входити струм різко зрушує Потенціал Спок фібробластів и вінікає МІП. Его амплітуда растет до того часу, поки НЕ почнет розслабленого міокарда, что веде до інактівації ціх каналів. Спадну фазу МІП обумовлює, мабуть, струм, який виникає за рахунок виходу з клітки K + через калієві канали, що активуються кальцієм. Але можливо, ця фаза пов'язана з інактивацією МСК.
Фібробласти специфічно, інакше, ніж кардіоміоцити, реагують і на штучну поляризацію мембрани, і на розтягнення тканини серця. Так, викликана гиперполяризация внутрішньої мембрани фібробластів спонтанно скорочується серцевого фрагмента зазвичай призводить до збільшення амплітуди МІП, а деполяризація - до зменшення. Примітно, що перший зрушення залежить від величини, на яку змістився потенціал спокою фібробластів, а другий - від ступеня штучної поляризації.
Додаткове розтягнення тканини спонтанно скорочується фрагмента правого передсердя викликає зрушення потенціалу спокою в сторону гіперполяризації мембрани фибробласта. Відповідно збільшується і амплітуда МІП, причому її величина прямо пов'язана зі ступенем гіперполяризації. Якщо розтягнення усунути, потенціал спокою повернеться до початкової величини. Настільки виражена реакція фібробластів на розтягнення свідчить, на наш погляд, про їхню участь в здійсненні механоелектричного зворотного зв'язку.
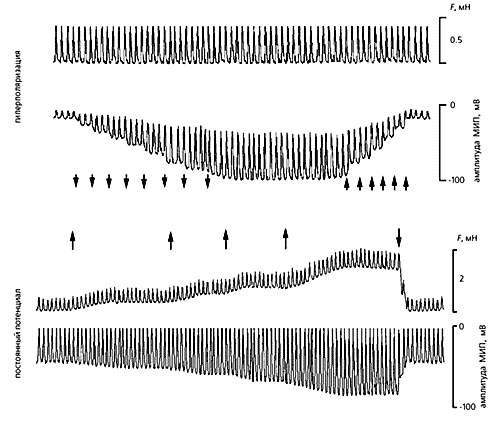
Реакції фібробластів з передсердя щура на штучну поляризацію мембрани і розтягнення клітини.Розтягнута тканина ритмічно скорочується (верхня механограмму) з силою близько 0.4 мН.Амплітуда потенціалу (МІП), індукованого таким впливом, збільшується на кожному кроці штучного посилення гіперполяризації мембрани (моменти посилення відзначені стрілками, спрямованими вниз) і зменшується, коли гиперполяризация так само поступово знімається (стрілки, спрямовані вгору).Якщо ж потенціал мембрани підтримувати на постійній величині, а силу розтягування збільшувати кілька разів, то в міру зростання сили, що розтягує амплітуда МІП теж збільшується.Все це підтверджує участь фібробластів в механічних і електричних події в серце.
Ролі строми (сполучної тканини, до складу якої входять фібробласти), в нормальному і хворе серце приділялося мало уваги. А тим часом ще в 1971 р описані випадки ішемії і інфаркту міокарда, що супроводжувалися запальним процесом тканини, який призвів до часткового або повного пошкодження кардіоміоцитів, але робота фібробластів активізувалася. Ці клітини перетворилися в міофібробласти з більшим ядром, рясним ендоплазматичним ретикулумом і Актинові філаменти. Наявність останніх структур цілком допускає думку, що такі трансформовані фібробласти здатні скорочуватися.
Ми вивчали електричні характеристики фібробластів правого передсердя щура після інфаркту (його викликали перев'язування коронарної артерії) міокарда і встановили, що потенціал спокою і опір мембрани залежали від розміру зони, охопленої інфарктом. Чим більше вона була, тим більше зміщувався потенціал спокою фібробластів: від -22 мВ у контрольних тварин і -25.8 у щурів з дрібновогнищевим інфарктом до -46.5 мВ - з великим.
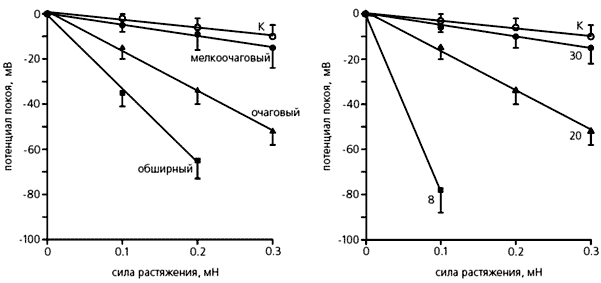
Зміна потенціалу спокою фібробластів при розтягуванні тканини правого передсердя щура з інфарктом міокарда лівого шлуночка.Зліва: зміни, що залежать від розміру інфарктного вогнища, праворуч - від кількості днів, що минули після інфаркту;К - контрольні вимірювання, проведені на фібробластах здорових щурів.Видно, що потенціал спокою знижується в міру збільшення вогнища хвороби, тобтоці "опорні" клітини стають більш чутливими до механічного впливу.Чітко помітна і підвищена чутливість фібробластів до розтягування тканини в ранні терміни після інфаркту.
Як можуть позначитися більш негативний потенціал спокою і підвищена амплітуда МІП фібробластів? На нашу думку, перший ефект здатний знизити спонтанну активність клітин водія ритму, а другий - викликати подовження потенціалу дії сусідніх кардіоміоцитів.
У тварин з різною величиною инфарктной зони відрізнялася також і чутливість фібробластів до механічного фактору. Примітно, що проявлялася та сама залежність, яку ми встановили в дослідах по вимірюванню електричних характеристик. Однак реакція фібробластів була пов'язана і з часом, що минув після інфаркту - чим менша кількість днів відділяло наступ хвороби від початку досліду, тим сильніше зсувається потенціал спокою фібробластів навіть при їх незначній розтягуванні.
Реакція ізольованого фибробласта на здавлювання (зліва) і розтягнення.Видно, що в першому випадку електрична провідність збільшується в порівнянні з нормальною клітиною (К), і чим більше вона стиснута, тим сильніше струм, але потенціал спокою при цьому зменшується.Відповідь фибробласта на розтягнення протилежний - його мембранна провідність знижується, а потенціал спокою збільшується.
Ми пояснюємо підвищену чутливість фенотипически змінених інфарктом фібробластів (т.е.міофібробластов) до механічного впливу збільшенням числа МСК в мембрані (їх участь підтверджується дією інгібітору каналів - іонами гадолінію). Навіть невелике розтягнення здатне активувати додаткову кількість МСК і тим підвищити вхідний струм. А оскільки зростає і концентрація вільного внутрішньоклітинного кальцію, відкриваються залежні від цього іона калієві канали і мембранний потенціал сильно зсувається в бік гіперполяризації.
Як же передаються потенціали між фібробластами і кардиомиоцитами? У серці фібробласти взаємодіють між собою переважно через високопроніцаемие контактні зони мембран, що містять кластери специфічних міжклітинних каналів. Вони різко знижують опір мембрани сусідніх клітин і роблять можливим їх електричне взаємодія. Такі ж канали, але розподілені дифузно, існують і в зоні контакту фібробластів з кардиомиоцитами. Зміна, скажімо, мембранного потенціалу перших клітин може викликати такий же відгук у сусідніх з ними кардиомиоцитах. Навколо клітин водія ритму в серці утворені кільця з фібробластів. Саме їх мембранний потенціал збільшується за абсолютною величиною і зрушує потенціал спокою кардіоміацітов. В результаті такого зміщення у клітин водія ритму зменшується частота виникнення потенціалу дії, а значить, і частота серцевих скорочень. Якраз це і спостерігається після інфаркту.
Щоб зрозуміти, як фібробласти відгукуються на механічний стимул (розтягнення або здавлювання), ми вимірювали струми, що протікають через мембрану ізольованих, одиничних клітин [ 8 ]. За результатами з'ясувалося, що здавлювання завжди призводить до зміщення потенціалу спокою в сторону деполяризації і реверсії вхідного струму на виходить. Чітко проявлялося і інгібуючу дію гадолінію. Далі слід було дізнатися, потоком катіонів або аніонів обумовлені виявлені ефекти. Для цього ми замінили іони K + на іони Cs + в умовах здавлювання клітини і виявили ту ж тенденцію, що і в попередніх експериментах: потенціал спокою зміщувався в бік деполяризації (при стисканні на 4 мкм він зміщувався від -32 мВ в середньому до -13 мВ), полярність диференційних струмів теж змінювалася. В експериментах з виявлення ролі аніонів Cl - ми не виявили їх впливу ні на потенціал мембрани, ні на струми. Отже, відповідь фибробласта на здавлювання здійснюється через катіонні потоки.
А що відбувається при розтягуванні цієї клітини? Виявилося, що воно призводить до гіперполяризації мембрани і значно зменшує силу струму в порівнянні з контролем. (Цікаво, що іони гадолінію діють ще сильніше, ніж механічний стимул.) Вплив аніонів хлору тут теж не було виявлено.
Таким чином, результати, отримані на ізольованих фибробластах і на багатоклітинних препаратах, збігаються. Цим ми довели своє вихідне припущення про те, що фібробласти серця відповідають на механічну стимуляцію зміною іонних струмів. Обумовлено це, як ми вважаємо, неселективной катионной провідністю. Оскільки механочувствітельние канали фібробластів реагують на напрям прикладеної сили (инактивируются розтягуванням і активуються внаслідок стискання), можна вважати, що скорочуються в систоле кардіоміоцити діють на лежать між ними фібробласти як біологічне здавлює пристрій.
Отже, кардіоміоцити і в ще більшому ступені фібробласти ефективно перетворять механічне подразнення в електричні відповіді, причому робота перших клітин модулюється другими. У здоровому серці розтягнення кардіоміоцитів, що приводить до деполяризації їх мембрани, і розтягнення фібробластів, що викликає гіперполяризацію, знаходяться в рівновазі. При патології же реакція на таке механічне подразнення виражена особливо сильно - але по-різному - в тих і інших клітинах. Якщо величина гіперполяризації фібробластів більше, ніж деполяризації кардіоміоцитів, серцевий ритм стає рідше і навіть може припинитися. І навпаки, якщо переважає остання, починається аритмія і може розвинутися фібриляція. Математичне моделювання повністю підтверджує такі висновки з експериментальних даних.
Виявлене нами взаємодія цих двох типів клітин і його вплив на роботу серця важливо не тільки для фундаментальної науки, а й для практичної медицини. Якщо врахувати, що повільне хронічне розтягнення міокарда за рахунок внутрисердечного тиску викликає зрушення потенціалів і як результат - порушення ритму, то можна зрозуміти, що навіть зміна пози хворого, який переніс інфаркт, може призвести до драматичних наслідків. Ми близькі до того, щоб перенести дослідження в клініку, але до цього треба буде вивчити електрофізіологічні характеристики кардіоміоцитів і фібробластів людських сердець - здорових і з різними патологічними змінами. Виходячи з наших уявлень про участь в механоелектричного зворотного зв'язку специфічних іонних каналів, необхідно знайти для них селективний блокатор, який переривав би цей зв'язок і тим відновлював нормальну роботу серця. Поки такого блокатора не існує. Але їм могли б стати антитіла до механочувствітельние каналах, правда, спочатку їх потрібно "клонувати". Можна блокувати одне з ланок того ланцюжка, яка призводить до експресії МСК, однак цей ланцюжок перш необхідно виявити. Пошуком блокаторів ми вже займаємося, і коли експерименти принесуть бажаний результат, не знадобляться ні електричний розряд, ні прямої механічний масаж, щоб вивести пацієнта зі стану фібриляції. Вона просто не виникне.
література
1. Камкін А.Г., Кисельова І.С., Яригін В.М. // Природа. 2002. №3. С.13-20.
2. Kamkin A., Kiseleva I., Wagner KD et al. // J. Mol. Cell. Cardiol. 2000. V.32. P.465-477.
3. Kiseleva I., Kamkin A., Wagner KDet al. // Cardiovasc. Res. 2000. V.45. P.370-378.
4. Камкін А.Г., Кисельова І.С. // Успіхи фізіолого. наук. 2000. Т.31. С.51-78.
5. Kamkin A., Kiseleva I., Isenberg G. // Cardiovasc. Res. 2000. V.48. P.409-420.
6. Камкін А.Г., Кисельова І.С., Яригін В.М. // Успіхи фізіолого. наук. 2001. т.32. С.75-104.
7. Камкін А.Г., Кисельова І.С. // Успіхи фізіолого. наук. 1998. Т.29. С.72-102.
8. Kamkin A., Kiseleva I., Husse B., Isenberg G. // Europ. J. Physiol. 2001. V.441. P.15-4, R191.
Як можуть позначитися більш негативний потенціал спокою і підвищена амплітуда МІП фібробластів?
Як же передаються потенціали між фібробластами і кардиомиоцитами?
А що відбувається при розтягуванні цієї клітини?