© В.М.Ройхель повільні хвороби
людини і тварин,
викликані пріонами
В.М.Ройхель
Віктор Мойсейович Ройхель, д. М. Н., Провідний науковий співробітник
Інституту поліомієліту і вірусних енцефалітів ім.М.П.Чумакова РАМН.
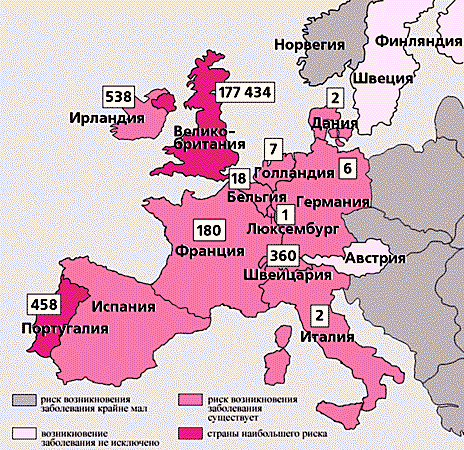
Ця група захворювань прославилася після вибуху в Великобританії епізоотії губкоподібної енцефалопатії великої рогатої худоби, більше відомої під назвою «коров'ячий сказ». До 1997 р було інфіковано близько 1 млн голів великої рогатої худоби, приблизно 54 тис. Інфікованих тварин потрапили в харчовий ланцюг людей [ 1 ]. Особливо був наляканий світ, коли з'явилися відомості про можливий зв'язок між цим захворюванням і виникненням нового варіанту хвороби Крейтцфельдта-Якоба - смертельної інфекції людини, яка, на відміну від її класичного варіанта, вражає молодих людей, як правило, у віці 27 років [ 2 , 3 ].
Немає нічого дивного, що ці події викликали підвищений інтерес фахівців до нового типу інфекції людини і тварин. Активізувалися пошуки, а потім вивчення вельми своєрідного збудника, принципово відрізняється від усіх відомих інфекційних агентів - віроідов, вірусів, бактерій і найпростіших. Хоча в Росії немає реальної загрози масового зараження збудником цього екзотичного захворювання, проте біомедичні дослідження ведуться і у нас.
"Історія хвороби"
Поняття «повільні хвороби» вперше ввів ісландський дослідник Б.Сігурдссон ще в 1954 році, який і сформулював основні характерні риси цієї особливої форми інфекції: ураження одного органу або однієї системи і наявність одного господаря, тривалий інкубаційний період (від кількох місяців до кількох років ), неухильненаростання клінічної симптоматики, неминуче приводить до смерті [ 4 ]. Сігурдссон вивчав повільні хвороби тільки тварин (зокрема, докладно дослідив поширене у всьому світі захворювання овець - скрепі), однак незабаром з'ясувалося, що вони можуть вражати і людини. У 1957 році американський вчений К.Гайдушек описав нове захворювання - куру, виявлене у папуасів-канібалів, жителів Нової Гвінеї. Хвороба носила масовий характер, і незабаром була доведена її інфекційна природа. За ці дослідження Гайдушек був удостоєний в 1976 р Нобелівської премії.
Збудниками повільних хвороб, як згодом з'ясувалося, можуть стати добре відомі «винуватці» гострих інфекцій: наприклад, віруси кору, краснухи, герпесу, грипу, кліщового енцефаліту та ін. (Всього близько 40 хвороб). Що станеться в організмі - повільне і фатальне протягом інфекційного процесу або гостре - залежить від умов, в яких опинився збудник [ 5 ].
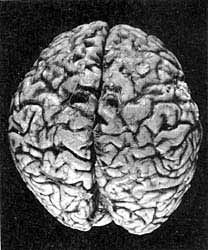
Головний мозок людини, яка загинула від хвороби Крейтцфельдта-Якоба. Явні патоморфологічні зміни: зменшення обсягу і маси мозку, витончення звивин півкуль великого мозку, переважно лобових і тім'яних доль зі значним розширенням борозен в цих областях. (В.А.Зуев, І.А.Завалішін, В.М.Ройхель, 1999.)
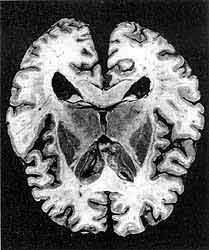
Горизонтальний зріз головного мозку людини, який помер від спорадичною форми хвороби Крейтцфельдта-Якоба. Захворювання призвело до звуження кори мозку в лобовій, тім'яній, скроневій і потиличній частках, а також відбулося деяке зменшення обсягу базальних ядер і таламуса і помірне розширення шлуночків мозку. (В.А.Зуев, І.А.Завалішін, В.М.Ройхель, 1999.)
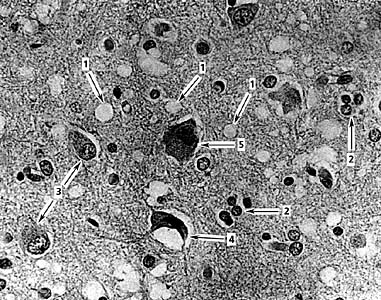
1 мікровакуолі, 2 - гинуть нейрони з гліальними вузликами, 3 - гіпертрофія астроцитів, 4 - зморщений нейрон, в якому зменшилися за обсягом цитоплазма і ядро, 5 - нейрон, в якому зібралося багато ліпофусцину і відбувся зсув ядра. Повів. ґ 400. (В.А.Зуев, І.А.Завалішін, В.М.Ройхель, 1999.)
Досить довго вважалося, що всі повільні хвороби викликаються вірусами, однак поступово, у міру накопичення фактичного матеріалу, стала виділятися особлива група захворювань людини і тварин, збудників яких стали називати «незвичайними вірусами». Своєрідність цих хвороб проявляється у виборчому ураженні центральної нервової системи, що неухильно призводить до Губкообразная станом сірого і / або білої речовини головного та спинного мозку при відсутності запальної реакції. З'ясування патоморфологічних і клінічних особливостей цих незвичайних захворювань дозволило виділити їх в окрему групу повільних хвороб під загальною назвою «трансмісивні Губкообразная енцефалопатії» (ТГЕ). За сучасною класифікацією, до них відносяться чотири хвороби людини і шість - тварин (табл.1).

Всі ці захворювання об'єднує наявність єдиного (або близького за властивостями) збудника, спільність патогенезу, експериментально показана можливість перенесення інфекції і відтворення клінічної та патогістологічної картини захворювання [ 6 ]. Ці висновки - результат багаторічних досліджень різних вчених - сформулював американський біохімік С.Прузінер, який отримав в 1997 р Нобелівську премію «за відкриття пріонів - нового біологічного принципу інфекцій». До цього всі спроби виявити збудників ТГЕ закінчувалися невдачею, хоча багато про їх властивості було відомо. Прузінер назвав інфекційний агент пріоном (часткова анаграма від англ. Proteinaceous infectious particles - белковоподобних інфекційна частка), а білок - РrP (від англ. Prion protein).
Пріони, дійсно, незвичні патогени, вони не здатні викликати гостру форму інфекції. Пов'язано це, мабуть, з повільним процесом «переродження» в зараженому організмі неінфекційного клітинного білка РrРС (С від англ. Cеll - клітина) - нормального компонента тканин ссавців, в тому числі і людини, - в інфекційний пріонних білок РrРSc (Sc від англ. scrapie - назви найбільш поширеною в природі пріонів інфекції овець і кіз).
Оскільки вже ні у кого не викликає сумнівів, що збудниками ТГЕ можуть бути тільки пріони, такі захворювання прийнято називати пріонні [ 7 ]. Практичний інтерес до цих смертельно небезпечних інфекцій безумовно пов'язаний з подіями останнього часу і збільшується ймовірністю зустрічі з цими хворобами. Теоретичний же інтерес до проблеми обумовлений результатами молекулярно-біологічних досліджень пріонів - нових і незвичайних збудників важких захворювань людини і тварин, які, як з'ясувалося, можуть виникати не тільки в результаті інфекції, але і спорадично і навіть передаватися у спадок.
Структура поранених білків
Над з'ясуванням структури і хімічної природи збудників ТГЕ працювало чимало фахівців протягом, принаймні, півстоліття. В результаті з'явилася велика кількість різноманітних гіпотез, багато з яких тепер мають лише історичне значення. Удача посміхнулася групі дослідників з Каліфорнійського університету (США), що працюють під керівництвом Прузінер. Головна їх заслуга в тому, що їм вдалося з'ясувати білкову природу пріонів. Розроблена американськими вченими багатоступенева система виділення вихідного інфекційного матеріалу дозволила отримати препарати, очищені в 100-1000 разів. Агент залишався стійким до впливу реагентів, инактивирующих нуклеїнові кислоти, що вказувало на їх відсутність в його складі. Вивчення очищеного препарату показало, що він має молекулярної масою близько або менше 50 кДа. В результаті подальшого очищення пріона з'ясувалося, що його основний компонент - мажорний білок з молекулярною масою 27-30 кДа, що позначається як РrР 27-30.
За фізико-хімічної характеристики РrР 27-30 - сіалоглікопротеін (олігосахарідсодержащій мембранний білок із залишками сіалова кислоти, які надають молекулі негативний заряд) і перший ідентифікований структурний компонент приона. Виявлення РrР 27-30 на етапі розвитку інфекції, тобто до появи патологічних змін в тканинах, - свідоцтво того, що цей білок не може бути вторинним продуктом патологічної реакції. Так стало очевидним, що РrР 27-30 грає центральну роль в патогенезі захворювання.
При подальшому вивченні пріонів, виділених з головного мозку заражених скрепі тварин, були виявлені частинки у вигляді стрижнів діаметром 10-20 нм і довжиною 100-200 нм. За ультраструктурі вони нагадували амілоїд (аномальний білок, який зазвичай утворюється при хронічних захворюваннях, наприклад туберкульозі легенів, кісток і т.д.) і, мабуть, являли собою полімерну форму пріона: кожен стрижень містив близько тисячі молекул пріона.
Важливим кроком, що має як теоретичне, так і методичне значення, було отримання антитіл при використанні в якості антигену високоочищених пріонів скрепі. У сироватках кроликів, яким вводили РrР 27-30, виявлені антитіла не тільки до нього, а й до інших білків, що відрізняється більш низькою молекулярною масою. Очевидно, ці білки або мають однакову антигенною детермінантою (областю антигену, комплементарної антитілу) з РrР 27-30, або вони - продукт його розщеплення. За допомогою виготовленої антисироватки з пероксидазною міткою вдалося виявити локалізацію пріонів в певних відділах головного мозку заражених тварин (табл.2). Згідно з раніше отриманими даними, структури, пов'язані з міченої антисироватки, володіли характеристикою амілоїдних бляшок. Використання антисироватки до синтетичного пептиду, відповідному N-кінцевій частині пріона, дозволило провести індикацію білка скрепі-асоційованих фібрил в головному мозку, селезінці і лімфатичних вузлах заражених тварин. При цьому позитивні результати були отримані на ранніх етапах інкубаційного періоду.

Визначення амінокислотноїпослідовності РrР 27-30 дозволило в 1985 р ідентифікувати кодує його ген Prnp. Виявилося, що цей ген міститься в геномах не тільки інфікованих скрепі тварин, а й здорових. Відповідно мРНК для РrРС була виявлена в головному мозку і в інших тканинах як інфікованих, так і контрольних тварин. Використовуючи відповідну антисироватки, вдалося показати, що в тканинах незаражених тварин міститься білок, споріднений РrР 27-30, але відрізняється від нього чутливістю до обробки протеазой К.
Були вивчені також деякі інші характеристики пріонів скрепі і хвороби Крейтцфельдта-Якоба. Зокрема, було підтверджено припущення про те, що інфекційна частка агента містить дві молекули РrР і що так звані сімейні форми (тобто зі спадковою схильністю) поранених захворювань пов'язані з конкретними мутаціями в гені Prnp. Наприклад, мутація, що викликає заміну проліну на лейцин в 102-м положенні РrР, виявилася пов'язана з розвитком синдрому Герстманна-Штреусслера-Шейнкера, а заміна аспарагінової кислоти на аспарагін (мутація в 178-м кодоні) може бути пов'язана як з хворобою Крейтцфельдта-Якоба , так і зі смертельною сімейної безсонням. До сьогоднішнього дня відомо вже про 20 мутаціях в гені Prnp людини, пов'язаних з сімейними формами пріонних захворювань.
Фізико-хімічні властивості
У другій половині 90-х років, коли вже була визначена амінокислотна послідовність РrР і виявлено ген Prnp, почалися інтенсивні пошуки причин патогенності пріонів. За допомогою сучасних методів молекулярно-генетичного аналізу були отримані нові дані про можливі варіанти складу і конформації (укладання) поліпептидного ланцюга РrР. Зокрема, було встановлено, що конверсія нормального пріонів білка в його інфекційну ізоформу - посттрансляційних процес [ 8 ]. Аналіз вторинної структури РrРSc показав, що цей перехід характеризується великими структурними змінами самого пріона. Клітинний білок містить 42% a -спіралей і майже не містить b -тяжей (всього близько 3%), в той час як в його інфекційної формі виявляється 30% a-спіралей і 43% b -тяжей [ 9 ]. В експериментальних дослідженнях було підтверджено, що обробка неінфекційного білка реагентами, що знижують утворення b -тяжей, також приводила до зменшення інфекційності переродженого приона. Одночасно знижувалася і стійкість РrРSc до дії протеази К, чутливість до якої вважається маркером, що відрізняє РrРC від РrРSc.
Перетворення нормального білка в патогенний, судячи з усього, відбувається шляхом білок-білкових взаємодій, при цьому не має значення, потрапляє РrРSс в організм ззовні або виникає в ньому спонтанно (в разі спонтанних і спадкових пріонних хвороб). Як це відбувається, впевнено сказати поки не можна, однак в даний час пропонуються дві моделі, які описують це перетворення: «гетеродімерная» і «полімеризації». Згідно з першою, пріони стан притаманне мономеру білка РrР, який каталізує конформаційний перехід молекули РrРC в форму РrРSс [ 10 ]. Після того як нормальний білок набуває пріонні властивості, димер дисоціює, і дві звільнилися молекули РrРSс можуть знову взаємодіяти з черговими молекулами РrРC. Процес нагадує ланцюгову реакцію і може протікати досить швидко, однак залишається непоясненим механізм утворення амілоїдних бляшок. Друга модель з цієї точки зору більш перспективна, оскільки розглядає пріон як упорядкований полімер РrР. А процес його конформаційні перебудови, згідно з цією теорією, нагадує кристалізацію, яку запускає олигомер РrРSс [ 11 ]. Відкладення білка РrРSс (бляшки) в тканинах мозку хворих людей зазвичай містять ниткоподібні агрегати цього білка, що свідчить про впорядкованої його полімеризації.
Таким чином, в результаті різносторонніх досліджень були отримані і систематизовані мають принципове значення дані про структуру та фізико-хімічні властивості пріонних білків. Аналіз цих відомостей створив необхідні передумови для подальшого поглибленого вивчення біологічних особливостей пріонних білків і механізму розвитку викликаються ними захворювань людей і тварин.
біологічні особливості
Незважаючи на велику кількість результатів у вивченні поранених білків, роль PrPC в живому організмі до кінця не відома. Ясно одне - цей білок життєво необхідний і еволюційно консервативний, оскільки виявлений у багатьох ссавців, птахів і навіть у нижчих евкаріот. Так, при аналізі первинної структури РrРС було виявлено, що 80% послідовностей РrРС у різних видів тварин ідентичні, виняток становив лише курячий РrРС, де ідентичність послідовностей по відношенню до інших видів становила всього 30%. Проте 24 амінокислотні послідовності, що розташовуються між 112-м і 135-м амінокислотними залишками, містяться в геномах всіх ссавців, а також курей.
В експериментах на трансгенних мишах, гомозиготних по втраті гена Prnp, було показано, що ці тварини після народження росли «нормальними», але через 70 тижнів у них розвинулися прогресуючі симптоми атаксії, зокрема порушилася моторна координація внаслідок екстенсивної втрати клітин Пуркіньє (великих нейронів кори мозочка). Крім цього, встановлено, що PrPC грає важливу роль в регуляції ціркадіанних (добових) ритмів, можливо, бере участь в активації лімфоцитів, а також виконує функції трофічного фактора для деяких популяцій нейронів. Збереження PrPC має значення для реалізації нормальної функції синапсів.
В останні роки з'явилися дані, що свідчать про роль клітинного білка в регуляції сну, і більш того - виникнення смертельної сімейної безсоння пов'язують з порушенням нормальної функції цього білка [ 12 ]. У дослідженнях in vitro було показано, що PrPC втягується в процеси регуляції вмісту внутрішньоклітинного Са 2+ в нейронах [ 13 ]. Уже доведеним можна вважати і значення нормального клітинного пріона в збереженні резистентності нейронів і астроцитів до окислювального стресу, і участь цього білка в метаболізмі міді в головному мозку [ 14 ]. А зовсім недавно були отримані дані про участь PrPC в трансдукції сигналів в нервовій тканині [ 15 ]. Цей список можна було б продовжити, але і так ясно, що уявлення про біологічну значущості PrPC в останні роки істотно розширилися.
Тепер стало відомо, что PrPC сінтезується в ендоплазматичної мережі и Досить Швидко деградує (всього за 5-6 годин). Сінтезованій PrPC, проходячи через апарат Гольджі, транспортується на поверхню Клітини, де ВІН зв'язується з глікофосфатіділінозітолом и надалі переноситися уздовж аксона с помощью Швидкого и активного (антероградного) транспорту. На Відміну Від PrPC інфекційній пріонніх білок ПЕРВИННА акумулюється в клітінах, накопічуючісь в цитоплазматичних везикулах. Подальше його накопичення в синаптичних структурах і пов'язана з цим дезорганізація синапсів, очевидно, стає причиною глибоких неврологічних дефектів і деменції.
У вивченні патогенезу і епідеміології пріонних хвороб існує багато білих плям. До них відносяться, зокрема, шляхи передачі захворювання в природі, взаємозв'язок хвороб людини і тварин, визначення «вхідних воріт» інфекції [ 16 ]. Судячи з усього, найбільш ймовірний шлях передачі захворювання - аліментарний. Правомірність цього висновку була побічно підтверджена, коли для викорінення куру на о.Новая Гвінея досить було заборонити звичай ритуального канібалізму. Доведено це було і в експериментах, в яких тваринам (в тому числі і приматам) згодовували високо інфіковані субстрати - тканини головного мозку хворих тварин.
Отримання нових даних дозволило зробити висновок, що пріонні хвороби - нейродегенеративні, у виникненні яких фундаментальну роль відіграють конформаційні зміни пріонів, а сам механізм розвитку хвороби безпрецедентний.
* * *
Результати досліджень, проведених в останні 10-15 років, дозволили з нових позицій підійти до питання про природу агентів ТГЕ, а сума отриманих нових знань про пріони послужила підставою для оптимістичного висловлювання Прузінер: «Ера чорного ящика біології скрепі і хвороби Крейтцфельдта-Якоба, можливо , добігає кінця". Хочеться сподіватися, що проблема повільних пріонних хвороб дійсно буде вирішена в недалекому майбутньому. Успіхи в цій області очевидні, про що свідчить хоча б те, що на протязі 20 років Нобелівський комітет двічі зазначав вчених за досягнення в одній і тій же області медико-біологічних досліджень.
література
1. Кемпбелл П.М. // Зап. біол., мед. і фармац. хімії. 1998. №4. С.34-40.
2. Hill A., Debruslais M., Joiner M. et al. // Nature. 1997. V.389. P.448-450.
3. Calza L., Manfredi R., Chiodo F. // Recenti Prog. Med. 2001. V.92. P.140-149.
4. Sigurdsson P. // British Veterinary J. 1954. V.110. P.341-354.
5. Зуєв В.А. Повільні вірусні інфекції людини і тварин. М., 1988.
6. Prion Biology and Diseases / Ed. SBPrusiner. NY, 1999..
7. Зуєв В.А., Завалишин І.А., Ройхель В.М. Пріонні хвороби людини і тварин. М., 1999. ..
8. Pan K., Baldwin M., Nguyen J. et al. // Proc. Natl. Acad. Sci. 1993. V.90. P.10926-10966.
9. Smith C., Collinge J. // Essay Biochem. 1995. V.29. P.157-174.
10. Cohen FE, Pan KM, Huang Z. et al. // Science. 1994. V.264. P.530-531.
11. Jarrett JT, Lansbury PT // Cell. 1993. V.73. P.1055-1058.
12. Tobler I., Deboer Т., Fisher M. // J. Neurosci. 1997. V.17. P.1869-1879.
13. Herms J., Tings Т., Dunker S. et al. // J. Neurobiol. Dis. 2001. V.8. P.324-330.
14. Brown D. // J. Brain Res. Bull. 2001. V.55. P.165-173.
15. Martins V., Mercadante A., Cabral A. et al. // Braz. J. Med. Biol. Res. 2001. V.34. P.585-595.
16. Ройхель В.М. Патогенез і діагностика деяких повільних пріонових нейроінфекцій: Автореферат на здобуття наукового ступеня доктора медичних наук. М., 1997. ..
17. Brown P. // J. Microsci Res. Tech. 2001. V.54. P.71-80.