Стресовий вплив сприймається корою головного мозку і передається в гіпоталамус, де виробляється кортикотропин-вивільняє гормон (CRH), стимулюючий гіпофізарні рецептори. Підсумком цього процесу є секреція кортикотропіну в плазму, стимуляція кортікотропінових рецепторів в адреналової області наднирників і викид кортизолу в кров. Вплив на гіпоталамічні кортизолової рецептори по типу зворотного зв'язку призводить до зниження вироблення CRH з метою підтримки гомеостазу (рис. 2).
В даний час отримано достатньо багато даних, які свідчать, що кортизол і CRH беруть участь в патогенезі депресій. При депресії підвищений вміст кортизолу в плазмі, підвищений рівень CRH в спинно-мозкової рідини і збільшений вміст РНК і білка переносника CRH в лимбических областях мозку (Merali Z., et al., 2004). Дексаметазоновий тест виявляє відсутність нормальної реакції придушення вироблення кортизолу майже у половини хворих найбільш важкими формами депресій (Carol BJ et al., 2007). Клінічний ефект тімоаналептіческой терапії супроводжується зникненням позитивної реакції на дексаметазоновий тест. Підвищення змісту моноамінових нейромедіаторів в синапсі також впливає на
гіпоталамо-гіпофізарно-адреналової вісь і призводить до зменшення наслідків тривалого стресу (Holsboer Е, 2000). Можливо, що антидепресивний ефект більше пов'язаний з корекцією явищ вторинного стресу, викликаного хворобливими депресивними переживаннями, ніж з прямим впливом на гіпотімно афект. Цим же механізмом можна пояснити широту клінічних ефектів більшості антидепресантів, включаючи лікування панічного розладу, посттравматичного стресового розладу, булімії, передменструального синдрому і обсесивно-компульсивного розладу.
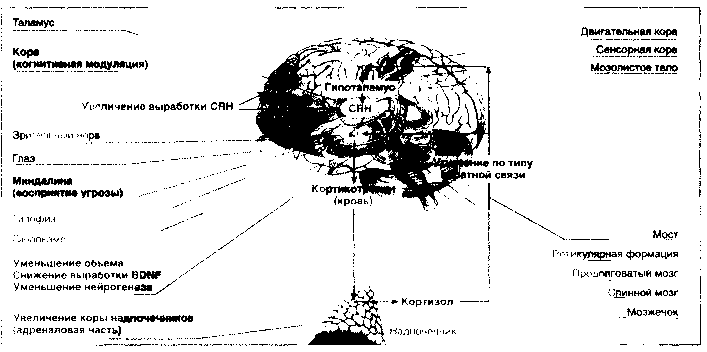
Мал. 2. Гипоталамо-гіпофізарно-кортизолової система при депресії
Примітка.
Гіпоталамо-гіпофізарно-кортизолової гіпотеза депресії постулює, що в її основі лежить порушення кортизолової реакції у відповідь на стресовий вплив. На схемі стрілками вказано, що на стрес реагують насамперед кора і мигдалина мозку, звідки сигнал передається в гіпоталамус, де відбувається вироблення кортікотропінвисвобождающего гормону (CRH), який спонукає секрецію кортико-тропіну в передній частині гіпофіза, який надходить безпосередньо в кров і стимулює адреналовую частина кори надниркових залоз і відповідну секрецію глюкокортикоидного гормону - кортизолу. Кортизол за механізмом зворотного зв'язку гальмує продукцію CRH і кортикотропіну відповідно в гіпоталамусі і в гіпофізі. Дослідження, що підтримують гіпоталамо-гіпофізарно-коргізоловую гіпотезу депресії, включають: підвищення рівня кортизолу в крові при важких депресіях; збільшення обсягу передньої частини гіпофіза і адреналової кори надниркових залоз; підвищення вмісту CRH в церебро-спінальної рідини; посилення експресії CRH в лимбических структурах; зменшення обсягу гіпокампу, числа нейронів і гліальних клітин, що, мабуть, пов'язано з пригніченням нейрогенезу, викликаного підвищеним вмістом кортизолу і зниженням вироблення мозкового нейротрофічних фактора (BDNF).
Антагоністи CRH-рецепторів показали обнадійливі результати в експериментах у тварин, проте не виявили виразної антидепресивний активності в клінічних умовах (Louis С., et al, 2006; Kehne JH, 2007; Binneman В. et al., 2008). При важких психотичних депресіях був зареєстрований ефект блокаторів глюкокортикоїдних рецепторів, зокрема міфепрістона (Flores В. Н. et al., 2006; De Battista C. et al., 2006; Beasey С. M. et al., 2009). Мифепристон також підсилює нейрогенез (Mayer JN et al., 2006) і підвищує чутливість 5-HT2A рецепторів в експериментальних моделях (Trajkovska V. et al., 2009).
Важливе значення в патогенезі депресій, очевидно, мають патологічні ціркадіанние коливання вмісту кортизолу в крові, особливо тривале підвищення рівнів кортизолу в нічний час, коли у здорових осіб він практично відсутній. Можливо також, що периферичний підйом кортизолу в крові лише відображає центральні порушення в системі CRH і його патологічну реакцію на тривалий стресовий вплив.
Підвищений вміст глюкокортикоїдів знижує нейропластічності мозку, включаючи здатність до нейрогенезу. При депресії зменшується обсяг гіпокампа (MacQueen G. М. et al., 2003). У посмертних дослідженнях у хворих на депресію були виявлені втрата клітин в поперековій звивині префронтальної кори, атрофічні зміни в дорсолатеральній префронтальної і орбитофронтальной корі, а також збільшення числа нервових клітин в гіпоталамусі і дорсальній частині ядра шва (центральна частина серотонинергической системи мозку) (Rajkowska G., 2000; Koolschijn Р. С. et al., 2009). Ці знахідки схожі на артофіческіе зміни в мозку при хворобі Кушинга, а також у експериментальних тварин, які перебували під впливом великих доз глюкокортикоїдів. Потрібно відзначити, однак, що при депресії підйом кортизолу в крові значно менше, ніж при хворобі Кушинга. Стрес пригнічує нейрогенез у тварин, включаючи приматів, а антидепресанти можуть запобігати придушення нейрогенезу в цих моделях і навіть стимулюють нейрогенез (Porera Т. D. et al., 2007). Клінічне значення цього феномену у хворих депресією залишається неясним.
Мозковий нейротрофічний фактор (BDNF) є головним нейротрофическим пептидом в організмі, відповідальним за процеси нейропластичности, включаючи зростання аксонів, збільшення числа синапсів і виживання клітин. Зміст BDNF знижується під впливом стресу і кортизолу (Angelucci Е et al., 2005; Kozlovsky N. et al., 2007). Посмертні дослідження мозку у жертв суїцидів, які страждали на депресію, виявили зниження концентрації BDNF в гіпокампі (Karege F. et al., 2005). Антидепресанти і ЕСТ збільшують вироблення BDNF і гормону росту (Chen В. et al., 2001). В одному з досліджень було показано зменшення обсягу гіпокампу у хворих депресією з аллелем BDNF по metl66 (Frodl Т. et al., 2007). Не виключено, що BDNF є сполучною ланкою між стресом, Нейрогенез і атрофією гіпокампа при депресії. Разом з тим, генетичні дослідження поліморфізму гена BDNF по vaIl66 met не виявлено зв'язку з депресією. Мабуть, BDNF є відносно неспецифічним механізмом, задіяним при різних психічних захворюваннях. Блокування гена BDNF у мишей не викликає депрессоподобной реакції (Lyons W. Е. et al., 1999), однак може знижувати ефект антидепресантів (Duman RS, Monteggia L. М., 2006). Запальні процеси і деякі нейротоксини також збільшують зміст BDNF в мозку (de Pablos R. М. et al., 2006). Хоча антидепресанти самостійно можуть викликати утворення нових синапсів, сприяючи більш ефективної переробки зовнішніх імпульсів (Castren Е., 2005), більшість з них змінюють експресію BDNF (Watanabe К. et al., 2010).
При стресової реакції за допомогою нейропептидів посилюється глутаматних нейротрансмісію, що було продемонстровано при депресіях (Zarate С. A. et al., 2004; Feyssa А. М. et al., 2009). Викид глутамату (гли) і збудливих нейропептидів збільшує нейротоксичність і веде до передчасного клітинному апоптозу. Антагоністи NMDA рецепторів (кетамін, рилузол і траксопріділ) дають швидкий, але короткочасний антидепресивний ефект при терапевтично резистентних депресіях (Sanacora G. et al., 2004; Zarate CA et al., 2006; Preskorn SH et el., 2008; Phepls LE et al., 2009). Нещодавно здатність до нормалізації глутаматергіческой трансмісії була виявлена у відомого антидепресанту з недостатньо зрозумілим механізмом дії тианептина (Mс Ewen В. S. et al., 2010).
Епідеміологічні дані вказують на те, що депресія часто передує серцево-судинних захворювань, ускладнює їх перебіг і призводить до підвищеної смертності (Lesperance F et al., 2007). У патогенезі як депресії, так і серцево-судинної патології бере участь ряд загальних факторів, зокрема, брак омега 3 жирних кислот (Nemets В. et al., 2002) і збільшення вмісту в плазмі гомоцистеїну (Folstein М. et al., 2007) . Підвищений рівень кортизолу в крові при депресії збільшує ризик ураження коронарних судин, оскільки кортизол сприяє розвитку вісцерального ожиріння (Brown Е. S. et al., 2004). Застосування антидепресантів збільшує виживання хворих з інфарктом міокарда (Taylor С. В. et al., 2005). При депресії і серцево-судинних захворюваннях описуються ознаки запального процесу (Muller N., 2006). Можливо, що в патогенезі депресії і серцево-судинних захворювань важливу роль відіграє ураження ендотеліальних клітин, які продукують BDNF і запускають процеси нейрогенеза.
Не менш важливу роль в патогенезі депресії, мабуть, грає і порушення імунної функції (Miller А. Н. et al., 2009). При депресії значно підвищено рівень прозапальних цитокінінів, таких як інтерлейкін6 (IL-6) і α-фактор некрозу пухлин (TNFα), який знижується у хворих з ефектом антидепресантів (Capuron L. et al., 2003; O'Brien S. М. et al., 2007; Hernandez ME et al., 2008). Зміст цих цитокінінів підвищено при багатьох соматичних захворюваннях, включаючи рак, і корелює з розвитком депресії при цих станах. Введення цих факторів тваринам викликає психомоторну загальмованість, зниження сексуальної активності і ангедонію (Anisman Н. et al., 2005; Janicki-Deverts D. et al., 2007). Кілька специфічних лікарських засобів - інфліксімаб (TNFα інгібітор) та целекоксиб (СОХ-2 інгібітор), в попередніх дослідженнях виявили антидепресивну активність (Akhondzaden S. et al., 2009 року; Muller N "2010).
Стресорні гормони, моноаміни та нейротрофічні чинники можуть впливати на ціркадіанние ритми, приводячи до розвитку депресії (Burke Н. М. et al., 2005; Ramirez-Rodriguez G. et al., 2009).
Якщо ви знайшли помилку, будь ласка, виділіть фрагмент тексту і натисніть Ctrl + Enter.